Edit detail for FoldingAndAliasing revision 1 of 3
| 1 2 3 | ||
|
Editor: damberger
Time: 2007/01/22 13:45:28 GMT+0 |
||
| Note: | ||
changed: - Folding and Aliasing Folding resp. Aliasing occurs when the spectral sweep width is smaller than the range of resonance frequencies in the spectrum. Spins outside of the sweep width are either *folded* into the spectrum (top->top) or *aliased* (top->bottom), depending on the experiment's method of frequency discrimination (TPPI vs. STATES respectively). In XEASY, this was taken into account by attributing a folding/aliasing attribute 'x' to each peak. This is an integer number (most often -1,0 or 1). The observed chemical shift 'c' is then translated into the real shift by calculating 'c=c+x*sw'. Not setting 'x' (or setting it wrongly) for a certain number of peaks is a very common cause of errors in chemical shift lists generated for a structure calculation. CARA does not use a folding/attribute. It only works with the real chemical shift. E.g. although a 3D 15N NOESY may be folded in the 15N dimension, the strips are found by navigating in an unfolded 15N-HSQC. In case the chemical shift is outside of the 15N sweep width of the 3D, the peak is still placed in the real position in the 2D. I.e. you must go outside the range of frequencies defined by the sweep width of the 3D to see the peak position. If the *show folded* option is selected you will also see the intensity from the folded resonance at the real position. Be aware that CARA needs to know the folding type of the spectrum (RSH, States-TPPI, ...). *uxnmr2xeasy* produces XEASY .param files, which set this attribute to NO. You will need to change this entry using a Text-editor to RSH or States-TPPI depending on the experiment. Step-by-step-Guide 1. Open a spectrum in any window e.g. MonoScope <br /><img src="folding1.gif" /> The arrow points to a group of aliased peaks (they have opposite sign to the other signals). 2. Select an expansion in the top part of the spectrum. You can move the display region a little bit outside of the spectral width by using *Control Click-Drag*. This shows the position where the "real" chemical shifts for this group of peaks lie. <br /><img src="folding2.gif" /> 3. Use *View->Show folded* to show a folded copy of the spectrum outside of the spectral width. <br /><img src="folding3.gif" /> <br /> You can now work with the peaks at their real positions. For example, if you pick these folded peaks, the real chemical shifts will be defined in the peaklist. A gray line indicates the spectral edge. *Fit to window* or *ww* will always bring you back to the unfolded spectrum region. BACK: ShortCuts NEXT: GlobalCursor "Tutorials index":Tutorials
Folding and Aliasing
Folding resp. Aliasing occurs when the spectral sweep width is smaller than the range of resonance frequencies in the spectrum. Spins outside of the sweep width are either folded into the spectrum (top->top) or aliased (top->bottom), depending on the experiment's method of frequency discrimination (TPPI vs. STATES respectively).
In XEASY, this was taken into account by attributing a folding/aliasing attribute x to each peak. This is an integer number (most often -1,0 or 1). The observed chemical shift c is then translated into the real shift by calculating c=c+x*sw. Not setting x (or setting it wrongly) for a certain number of peaks is a very common cause of errors in chemical shift lists generated for a structure calculation.
CARA does not use a folding/attribute. It only works with the real chemical shift. E.g. although a 3D 15N NOESY may be folded in the 15N dimension, the strips are found by navigating in an unfolded 15N-HSQC. In case the chemical shift is outside of the 15N sweep width of the 3D, the peak is still placed in the real position in the 2D. I.e. you must go outside the range of frequencies defined by the sweep width of the 3D to see the peak position. If the show folded option is selected you will also see the intensity from the folded resonance at the real position.
Be aware that CARA needs to know the folding type of the spectrum (RSH, States-TPPI, ...). uxnmr2xeasy produces XEASY .param files, which set this attribute to NO. You will need to change this entry using a Text-editor to RSH or States-TPPI depending on the experiment.
Step-by-step-Guide
- Open a spectrum in any window e.g. MonoScope?
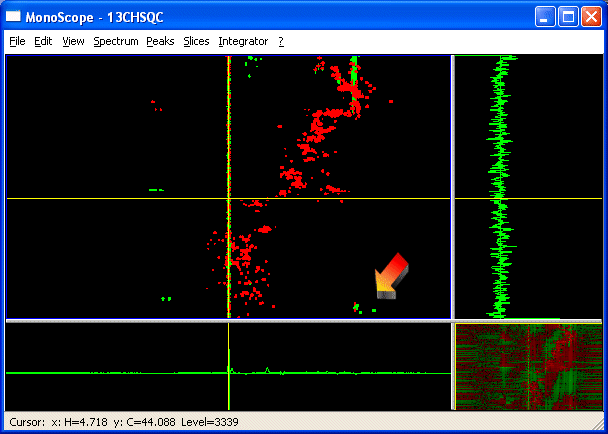
The arrow points to a group of aliased peaks (they have opposite sign to the other signals).
- Select an expansion in the top part of the spectrum. You can move the display region a little bit outside of the spectral width by using Control Click-Drag. This shows the position where the "real" chemical shifts for this group of peaks lie.
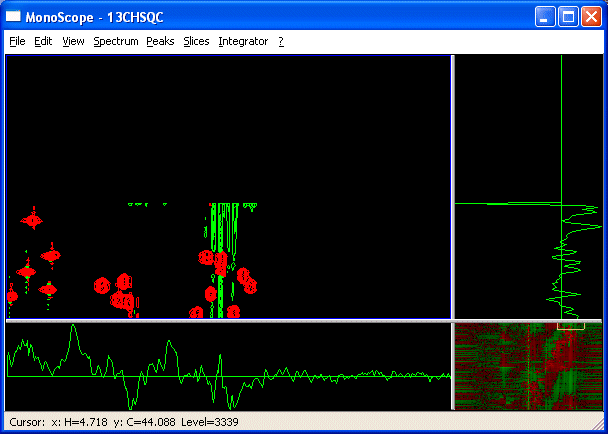
- Use View->Show folded to show a folded copy of the spectrum outside of the spectral width.
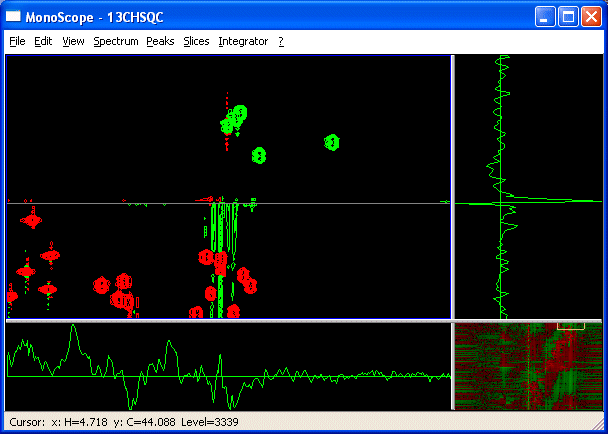
You can now work with the peaks at their real positions. For example, if you pick these folded peaks, the real chemical shifts will be defined in the peaklist. A gray line indicates the spectral edge. Fit to window or ww will always bring you back to the unfolded spectrum region.
BACK: ShortCuts?
NEXT: GlobalCursor?