Edit detail for ResidueType revision 1 of 1
| 1 | ||
|
Editor: damberger
Time: 2007/01/22 13:46:10 GMT+0 |
||
| Note: | ||
changed: - Residue Types are !CARAs way of modeling the structure of individual building blocks of a biological polymer. These can be amino acid residues, nucleotide residues of RNA or DNA, saccharide residues from oligosaccharides or any other building block which has a fixed linkage and builds a linear chain. CARA uses the topology or bond connections between atoms in the ResidueType to simulate the result of experiments ("SpectrumTypes":CreatSpectrumTypes). Expanded view of ResidueType ALA viewed in the ResidueType Explorer. From the standard template Start1.2.cara: <img src="ResidueTypeAla.gif" /><br> ResidueType contains the following type of information: Nomenclature 1. Full name of the residue. E.g. for Ala "Alanine". 2. Short name of the residue. E.g. for Ala "ALA". This is the actual Id of the residue and must be unique. 3. Letter name of the residue. E.g. for Ala "A". This is the label which appears next to peaks to indicate residue type. It can be more than one letter in length. Atoms 1. Names and Atom Type of each atom within a residue. E.g. Ala Methy group has !AtomType "H" and Name "HB". 2. Magnitude (number of equivalent spins) for each atom. E.g. Ala Methyl group has magnitude 3. 3. The average chemical shift (mean) and deviation of the atom. E.g. Ala Methy has mean=1.37, dev=1.00 (dev is the allowed range for matching NOT the standard deviation. It usually is 4 x SD). 4. x,y Position of Atom in the !ModelViewer (just cosmetic). 5. Group. Each atom can belong to a group which is a label that indicates a pseudoatom (when the chemical shift of the atoms belonging to the group is degenerate then this label is used in the assignment). Right-clicking on the ResidueType ALA and selecting "!MoleculeViewer" displays the ResidueType topology: <img src="MoleculeViewerAla.gif" /><br> Topology 1. connections (bonds) between atoms. (single and double bonds are not distinguished) E.g. Ala "HB" is connected to "CB". 2. Nterminal. Name of atom connecting to the previous residue in the polymer. (The direction N->C is a convention taken from amino acids but in Nucleic Acids for example Nterminal is "C5'") E.g. Ala "N". 3. Cterminal. Name of atom connecting to the next residue in the polymer. E.g. Ala "C". 4. Set Generic Residue. Only one residue is the generic residue. This is used to model the topology of the previous and next residue when the fragment is not yet placed in the sequence. 5. Spin System Type. Each residue can be included in a class of Spin System Types. Each Spin System Type can have its own Nterminal and Cterminal atom. One of the Residue Types in the Spin System class is defined as the model for that Spin System Type. When the user selects the Spin System Type in the "Pick New System" menu, the Residue Type which is model for the Spin System Type is used to generate the list of labels. The settings for Nterminal, Cterminal, Generic Residue are accessed by right-clicking on the ResidueType and selecting the appropriate menu item: <img src="ResidueTypeContextMenu.gif" /><br> BACK: "Creating a Spectrum Type":CreateSpectrumType Next: "Building a new ResidueType":CreateResidueType "Tutorials index":Tutorials
Residue Types are CARAs way of modeling the structure of individual building blocks of a biological polymer. These can be amino acid residues, nucleotide residues of RNA or DNA, saccharide residues from oligosaccharides or any other building block which has a fixed linkage and builds a linear chain. CARA uses the topology or bond connections between atoms in the ResidueType to simulate the result of experiments (SpectrumTypes).
Expanded view of ResidueType ALA viewed in the ResidueType Explorer.
From the standard template Start1.2.cara:
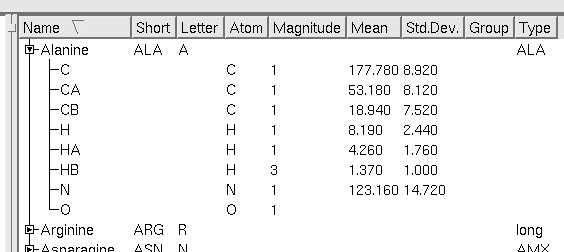
ResidueType contains the following type of information:
Nomenclature
- Full name of the residue. E.g. for Ala "Alanine".
- Short name of the residue. E.g. for Ala "ALA". This is the actual Id of the residue and must be unique.
- Letter name of the residue. E.g. for Ala "A". This is the label which appears next to peaks to indicate residue type. It can be more than one letter in length.
Atoms
- Names and Atom Type of each atom within a residue. E.g. Ala Methy group has AtomType "H" and Name "HB".
- Magnitude (number of equivalent spins) for each atom. E.g. Ala Methyl group has magnitude 3.
- The average chemical shift (mean) and deviation of the atom. E.g. Ala Methy has mean=1.37, dev=1.00 (dev is the allowed range for matching NOT the standard deviation. It usually is 4 x SD).
- x,y Position of Atom in the ModelViewer (just cosmetic).
- Group. Each atom can belong to a group which is a label that indicates a pseudoatom (when the chemical shift of the atoms belonging to the group is degenerate then this label is used in the assignment).
Right-clicking on the ResidueType ALA and selecting "MoleculeViewer" displays the ResidueType topology:
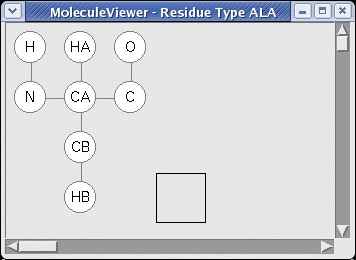
Topology
- connections (bonds) between atoms. (single and double bonds are not distinguished) E.g. Ala "HB" is connected to "CB".
- Nterminal. Name of atom connecting to the previous residue in the polymer. (The direction N->C is a convention taken from amino acids but in Nucleic Acids for example Nterminal is "C5'") E.g. Ala "N".
- Cterminal. Name of atom connecting to the next residue in the polymer. E.g. Ala "C".
- Set Generic Residue. Only one residue is the generic residue. This is used to model the topology of the previous and next residue when the fragment is not yet placed in the sequence.
- Spin System Type. Each residue can be included in a class of Spin System Types. Each Spin System Type can have its own Nterminal and Cterminal atom. One of the Residue Types in the Spin System class is defined as the model for that Spin System Type. When the user selects the Spin System Type in the "Pick New System" menu, the Residue Type which is model for the Spin System Type is used to generate the list of labels.
The settings for Nterminal, Cterminal, Generic Residue are accessed by right-clicking on the ResidueType and selecting the appropriate menu item:
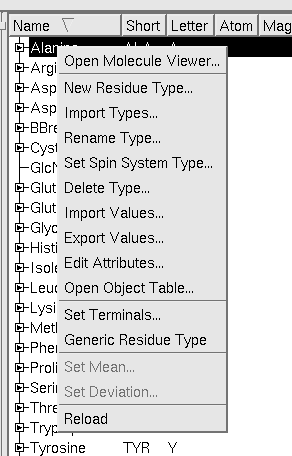
BACK: Creating a Spectrum Type